Informaciones Psiquiátricas - Primer trimestre 2002. Número 167
Base genética de las demencias degenerativas
Elisabet Vilella
Hospital Psiquiàtric Universitari Institut Pere Mata.
Unitat de Psiquiatria i Psicologia Mèdica.
Facultat de Medicina i Ciències de la Salut. Universitat Rovira
i Virgili.
Reus. Tarragona.
Lídia Figuera
Institut de Recerca en Ciències de la Salut (IRCIS).
Recepción: 18-02-02 / Aceptación: 11-03-02
La década de los 90 científicamente será recordada como la «década del cerebro» pero la primera edad de oro en el estudio de las enfermedades neurodegenerativas se remonta a principios del siglo xx, cuando Alzheimer (1907), Pick (1906) y Lewy (1912), gracias a los avances tecnológicos del momento, como el uso del microscopio y la tinción de tejidos, clasificaron las enfermedades neurodegenerativas como entidades clínicopatológicas. La clasificación actual de las demencias se esquematiza en la tabla I.

La investigación de las bases moleculares de la demencia se ha dirigido mayoritariamente a las demencias degenerativas y entre éstas, a las que afectan a mayor porcentaje de la población. Estas demencias son la enfermedad de Alzheimer, la demencia frontotemporal y la demencia con cuerpos de Lewy1.
DEMENCIAS PRIMARIAS DEGENERATIVAS MÁS FRECUENTES
Enfermedad de Alzheimer
La mayoría de las demencias son de origen degenerativo y la enfermedad de Alzheimer (EA) es la más frecuente de ellas representando un 60-75% del total de las demencias y afecta entre el 5-10% de la población mayor de 65 años. Su prevalencia aumenta con la edad siendo del 3% en la población de entre 65-74 años, 19% entre 75-84, alcanzando el 47% en la población de más de 85 años.
La EA puede clasificarse en función de la edad de aparición de los primeros síntomas y la existencia de antecedentes familiares. Así, esta enfermedad puede tener un debut anterior a los 65 años y hablaremos de EA presenil o de inicio precoz (EAEIP) o bien puede manifestarse a partir de los 65 años considerándose EA senil o de edad de inicio tardío (EAEIT). Esta enfermedad puede presentarse de forma familiar, cuando existen antecedentes en la familia del paciente o bien de forma esporádica cuando no se conocen antecedentes familiares. Estas clasificaciones no son excluyentes, así podemos encontrar casos familiares de edad de inicio tardío, o bien casos esporádicos preseniles.
Demencia con cuerpos de Lewy
Es la segunda demencia en frecuencia y representa entre el 10-25% del total de las demencias. La demencia con cuerpos de Lewy (DCL) suele manifestarse entre los 60 y 80 años aunque la edad de inicio de los casos descritos oscila entre los 20 y 90 años.
Demencia frontotemporal
Se trata de un grupo de enfermedades que representa entre el 12-20% del total de las demencias y el 20% de las demencias preseniles. Los primeros síntomas aparecen entre los 45-60 años y la mitad de los casos presentan antecedentes familiares. El arquetipo de la demencia frontotemporal es la enfermedad de Pick.
LESIONES CARACTERÍSTICAS DE LAS DEMENCIAS DEGENERATIVAS
En la actualidad, nos encontramos en la segunda edad de oro en el estudio de las enfermedades neurodegenerativas, coincidiendo con el progreso tecnológico de la genética y la bioquímica moleculares y de las técnicas de neuroimagen. La introducción de estas técnicas ha permitido la descripción molecular de las lesiones cerebrales descritas hace 100 años e identificar el componente genético de estas enfermedades.
Las demencias neurodegenerativas tienen en común la presencia de agregados proteicos insolubles que causan las lesiones que las caracterizan. Las proteínas de estos agregados, de las que hablaremos en detalle a continuación, ha sugerido una clasificación molecular de las demencias que queda resumida en la tabla II2.

Placas seniles
Las placas seniles son depósitos congofílicos extracelulares en los que el principal componente es el péptido b-amiloide (bA). El bA, es un péptido de 40- a 42-aminoácidos, que manifiesta una fuerte tendencia a agregarse y que proviene de la proteólisis de la proteína precursora del péptido b-amiloide (PPA). La PPA es una proteína transmembrana cuya función se desconoce y que es procesada mediante distintas proteasas (figura 1).
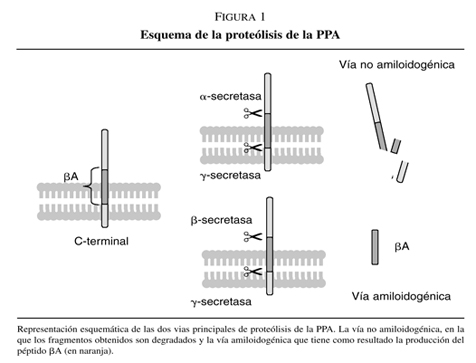
La vía de proteólisis más común es la de la acción de una proteasa, conocida como a-secretasa, que corta la proteína generando un fragmento extracelular y soluble. Posteriormente actuará una g-secretasa que libera la parte carboxilo-terminal al interior de la célula donde es degradado (ver figura 1). Esta vía previene la formación del bA. La segunda vía de proteólisis, conocida como vía amiloidogénica, se da cuando la primera proteasa que escinde la proteína es una b-secretasa que genera un fragmento C-terminal más largo y que tras la acción de la g-secretasa (ver figura 1), libera al espacio extracelular el bA. La alta hidrofobicidad de bA le lleva a formar autoagregados que constituyen las fibrillas insolubles que se encuentran en los depósitos amiloides3.
Estas lesiones son características de la enfermedad de Alzheimer y su presencia es absolutamente necesaria para el diagnóstico post-mortem de esta enfermedad, además la formación de las placas seniles o amiloideas precede a los síntomas de la enfermedad. Es por ello que las nuevas estrategias terapéuticas se dirigen hacia la prevención de la formación del bA, y en el punto de mira de los equipos de investigación se encuentran los compuestos inhibidores de la g-secretasa4. En la misma línea estaría la inmunización con bA, una «vacuna» que evitaría la formación de la placa senil y retrasaría los síntomas de la enfermedad5.
Nódulos neurofibrilares
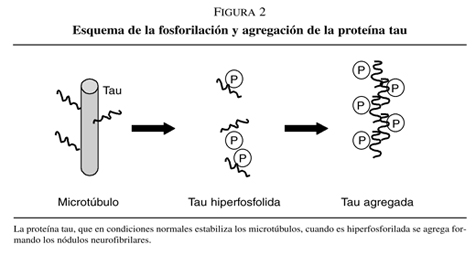
Los nódulos neurofibrilares están constituidos por precipitados intracelulares de filamentos de proteína tau.
En condiciones normales la proteína tau se asocia a los microtúbulos neuronales estabilizándolos y se le atribuyen además las funciones de regular el transporte de vesículas a nivel intracelular, facilita el crecimiento axonal y sirve de anclaje para determinadas enzimas. Cuando la proteína tau se fosforila forma filamentos helicoidales apareados pero si sufre una hiperfosforilación estos filamentos se agrupan formando los nódulos neurofibrilares que son altamente insolubles y precipitan en el interior de la célula (figura 2). En condiciones patológicas, cuyo origen se desconoce, la hiperfosforilación de la proteína tau tiene como consecuencia una desestabilización de los microtúbulos y como consecuencia una alteración del transporte axonal afectando la supervivencia de la neurona6.
Cuerpos de Lewy
Los cuerpos de Lewy están formados por agregados de a-sinucleína. Esta proteína pertenece a la familia de las sinucleínas y se concentra en los terminales nerviosos. Es abundante en el cerebro pero sus funciones fisiológicas son poco conocidas. Los mecanismos de agregación de esta proteína en las enfermedades neurodegenerativas, tampoco son conocidos7.
Estas lesiones son características de la demencia con cuerpos de Lewy y de la enfermedad de Parkinson, aunque están presentes en otras demencias como la enfermedad de Alzheimer.
CÓMO ABORDAR LOS ESTUDIOS GENÉTICOS DE LAS ENFERMEDADES COMPLEJAS
Las demencias, son enfermedades complejas o multifactoriales, resultado de la interacción entre factores ambientales y múltiples genes. Para detectar regiones genéticas específicas y genes involucrados en estas enfermedades se utilizan dos métodos complementarios, los estudios del análisis de ligamiento y los estudios de asociación8.
Estudios del análisis de ligamiento: Se basan en el fenómeno de recombinación que se da entre cromátidas homólogas durante la meiosis, de manera que cuando dos loci se encuentren muy próximos en un cromosoma la recombinación entre ellos es poco probable y por consiguiente se transmitirán juntos a la siguiente generación.
La muestra de estudio en estos casos la constituyen familias en las que la enfermedad se presenta en dos o más parientes de primer grado y nos ofrecen como resultado una aproximación a una región cromosómica candidata a contener el gen o genes causantes de la enfermedad de estudio (figura 3).ç
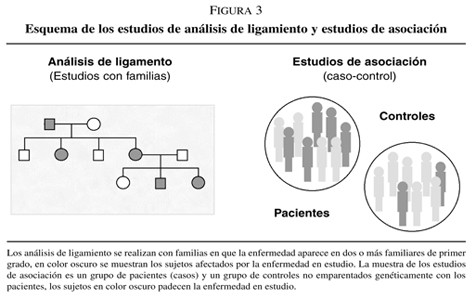
Estudios de asociación: Estos estudios también reciben el nombre de estudio de genes candidatos, puesto que los investigadores a priori, deciden el gen que van a estudiar basándose en su hipótesis de trabajo. En estos estudios se comparan las frecuencias alélicas de un polimorfismo del gen candidato escogido en dos poblaciones, una afectada por la enfermedad en estudio y otra control (no afectada). Estas poblaciones deben pertenecer al mismo grupo étnico y los sujetos de estudio no pueden estar emparentados genéticamente.
La comparación de las frecuencias entre las dos poblaciones puede manifestar una asociación positiva entre el polimorfismo y la enfermedad que será interpretada en términos de riesgo relativo a desarrollar la enfermedad. Este valor nos indica cuantas veces es mas frecuente la enfermedad en individuos que presenten un alelo determinado respecto a los que no lo presentan.
GENES CAUSANTES DE DEMENCIA
Gen PPA
Las placas seniles, características de la enfermedad de Alzheimer, se encuentran también en el cerebro de sujetos con el síndrome de Down. Este dato dirigió los estudios de análisis de ligamiento hacia el cromosoma 21, donde en 1987 se localizó al gen que codifica la PPA.
Las 13 mutaciones descritas en este gen se localizan en la región de la proteína que contiene el péptido bA y tienen como consecuencia un aumento de la producción de este péptido9. Estas mutaciones son claramente causa de la enfermedad de Alzheimer, pero tan solo en un 5-10% de las formas familiares de esta enfermedad.
Gen PS1
En 1995 se descubrió el gen PS1 (presenilina-1) en el cromosoma 14, causante del 30-50% de la forma presenil de la enfermedad de Alzheimer10. La proteína ps1 presenta 8 dominios transmembrana y su función no es conocida, aunque estudios con ratones transgénicos, ponen en evidencia que es un gen fundamental durante el desarrollo ya que los animales a los que se ha eliminado el gen, no sobreviven al primer día de vida y presentan malformaciones en su esqueleto11.
Los estudios genéticos realizados en familias afectadas de EA, hasta el momento han descrito 72 mutaciones en el gen PS1 distribuidas en todos los dominios de la proteína. Esta proteína está implicada en el desarrollo de esta enfermedad, aumentando la producción del péptido bA, mediante la activación de la g-secretasa si bien estudios recientes señalan que la propia ps1 podría ser la g-secretasa3.
Gen PS2
El tercer gen responsable la EA presenil familiar, es el PS2 que codifica para la proteína ps2 (presenilina 2) y se localiza en el cromosoma 1. Esta proteína presenta un 80% de homología con la ps1, y se han descrito mutaciones en el gen PS2 pero en número menor que PS1. En relación con la EA se le atribuye la función de aumentar los niveles de péptido bA11.
Gen TAU
En la literatura se han descrito una serie de familias que presentan una demencia frontotemporal con parkinsonismo, históricamente llamada enfermedad de Pick y que se transmite de una generación a la siguiente de forma autosómica dominante. Estudios con estas familias han descrito ligamiento de esta enfermedad con el cromosoma 17 por lo que se conoce a esta demencia con las siglas DFTP-17 (demencia frontotemporal y parkinsonismo ligada al cromosoma 17)12.
Los análisis histológicos de tejido cerebral de los pacientes afectados de DFTP-17 describen la presencia de nódulos neurofibrilares, el componente principal de los cuales es la proteína tau, como hemos visto anteriormente. El gen que codifica para esta proteína se encuentra en el cromosoma 17 y se han descrito 14 mutaciones, 9 de las cuales serían causantes de la DFTP-17. Estas mutaciones tienen como consecuencia una disfunción de la proteína que favorece la formación de los nódulos neurofibrilares característicos de las taupatías13.
Gen SNCA
El componente proteico de los cuerpos de Lewy es la a-sinucleína, cuyo gen, SNCA mapa en el cromosoma 4 y se han descrito 2 mutaciones causantes de la enfermedad de Parkinson.
GENES DE RIESGO PARA LA DEMENCIA
En el apartado anterior se describen mutaciones descritas en genes causantes de demencia pero que en realidad explican un porcentaje muy pequeño de estas enfermedades, así por ejemplo las mutaciones en los genes causantes de la enfermedad de Alzheimer explican de un 4 a 8% del total de los casos. Por lo tanto deben existir otros genes aún por identificar que expliquen el sustrato genético de muchos casos de demencia en los que los genes que acabamos de describir no están mutados. En la búsqueda de otros genes causantes, los investigadores han identificado otros genes que no pueden considerarse causantes pero sí de riesgo. A continuación hablaremos de los genes de riesgo más importantes para la demencia.
Gen APOE
La apolipoproteína E (apoE), es esencial para el transporte de colesterol en la circulación sistémica y entre células. El principal órgano de síntesis es el hígado pero se expresa en otros órganos como el cerebro donde participa en la regeneración neuronal y la plasticidad sináptica14.
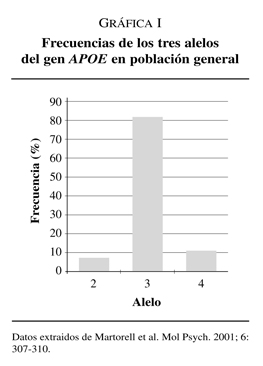
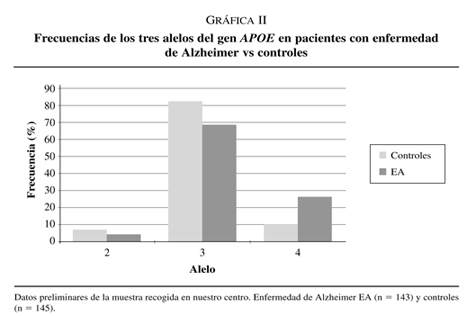
La apoE se presenta en tres isoformas mayoritarias, E2, E3 y E4 codificadas por 3 alelos e2, e3 y e4 de un único locus que mapa en el cromosoma 19q. El alelo e3 es el más común con una frecuencia del 78% de la población, el alelo e4 lo presenta el 15% de la población mientras que e2 tiene una frecuencia del 7% en población general (gráfica I)15.
En el año 1993 el grupo del Dr. Allen Roses describió una asociación positiva entre el alelo e4 del gen APOE y la enfermedad de Alzheimer de edad de inicio tardío16. Este resultado se ha replicado en múltiples estudios estableciéndose que el alelo e4 del gen APOE es un factor de riesgo para padecer la enfermedad de Alzheimer, además el riesgo es dosis dependiente aumentando casi 3 veces por cada alelo e4 que presenta el sujeto17.
El alelo e4, además está relacionado con la edad de inicio de la enfermedad de manera que los pacientes homozigotos para este genotipo (e4/e4) presentan los síntomas de la enfermedad antes que los pacientes heterozigotos (e3/e4)18. La diferencia de este gen con los genes que causan demencia es que el hecho de presentar uno o dos alelos e4, no significa que vaya a aparecer la enfermedad, así existen sujetos con los dos alelos e4 que llegan a los 90 años sin ningún síntoma de demencia y pacientes sin ningún alelo e4 que padecen la enfermedad de Alzheimer. Estas diferencias podrían explicarse no sólo con el genotipo del APOE sino con la concentración de la isoforma apoe4 en el cerebro. En esta línea existen estudios que demuestran una sobreexpresión del alelo e4 en el cerebro de pacientes que padecen la enfermedad de Alzheimer respeto a sujetos controles19.
Cromosoma 12
Como ya se ha comentado anteriormente, la forma de EA más común se presenta de forma esporádica y después de los 65 años de edad y para estas formas no se conocen genes causales, tan solo el gen APOE como factor de riesgo pero se estima que contribuye en un 50% de los casos seniles. Por esta razón los equipos de investigación continúan la búsqueda de nuevos genes implicados en el desarrollo de la enfermedad. En esta línea se ha identificado el cromosoma 12, en el que mapan dos genes, el A2M que codifica para la a-2-macroglobulina (a2m) y el LRP que codifica para el receptor LRP (low-density lipoprotein-related receptor protein)20.
LRP es un receptor multiligando que en la membrana de las neuronas reconoce e internaliza proteínas tan diferentes como apoE, PPA, y a2m. La proteína a2m es un inhibidor de proteasas que se une de forma muy ávida al péptido bA, el complejo a2m -bA es captado por el receptor LRP que lo internaliza para que sea degradado contribuyendo así a la eliminación del péptido bA extracelular y por lo tanto impidiendo la formación de la placa senil21.
Se han realizado numerosos estudios de asociación entre polimorfismos descritos en estos dos genes LRP y A2M y la enfermedad de Alzheimer pero los resultados obtenidos no son concluyentes22.
OTROS GENES
En la década de los 90 se realizaron numerosos estudios en busca de nuevos genes candidatos basándose en las hipótesis biológicas de la enfermedad, como los genes que codifican para otros receptores de la apoE (VLDLR y LDLR), proteínas que se encuentran presentes en la placa senil como a-1-antiquimotripsina (ACT), butirilcolinesterasa (BCHE-K) e interleuquina 1 (IL1A), receptores de los estrógenos (ER-a), la neurotrofina-3 (NT3), el receptor de serotonina (5HTR), genes relacionados con el estrés oxidativo como la sintasa de óxido nítrico (NOS3), entre otros23. Los estudios con estos genes que han dado resultados positivos en algunas poblaciones no han podido ser confirmados por otros autores al repetirlos en sus poblaciones de estudio. Por ello se estima que estos genes si tienen un efecto sobre la enfermedad, éste sería muy pequeño.
ÚLTIMAS CONSIDERACIONES
A pesar de la heterogeneidad de resultados obtenidos en el estudio de las bases moleculares de la demencia, los equipos de investigación no deben sino que aunar esfuerzos en conocer las causas de la enfermedad para poder desarrollar tratamientos farmacólógicos precisos y poder prevenir estas enfermedades.
BIBLIOGRAFÍA
1. Lowe J, et al. Non-Alzheimer degenerative dementias. Brain Pathology. (1998) 8: 295-297.
2. Hardy J, et al. Genetic clasification of primary neurodegenerative disease. Science. (1998) 282: 1075-1079.
3. Neve R, et al. Alzheimer’s disease: a re-examination of the amyloid hypothesis. Trends Neurosci. (1998) 21: 15-19
4. Dingwall C. Spotlight on BACE: The secretases as targets for treatment in Alzheimer disease. J. Clin. Invest. (2001) 108: 1243-1246.
5. Schenk DB, et al. b- Peptide Immunization. A possible new treatment for Alzheimer disease. Arch. Neurol. (2000) 57: 934-936.
6. Johnson G et al. Tau protein in normal and Alzheimer’s disease brain. Alzheimer’s disease review. (1996) 1: 34-54.
7. Lippa CF, et al. a-Synuclein in familial Alzheimer disease. Arch. Neurol. (2001) 58: 1817-1820.
8. Thomson G, et al. The genetics of complex diseases. Trends Genet. (1999) 15: M17-M20.
9. Lendon C, et al. Exploring de etiology of Alzheimer disease using molecular genetics. JAMA. (1997) 277: 825-831.
10. Sherrington R, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. (1995) 375: 754-760.
11. Czech C, et al. Presenilins and Alzheimer’s disease: biological functions and pathogenic mechanisms. Progress in Neurobiology. (2000) 60: 363-384.
12. Lee V, et al. Neurodegenerative tauopathies: human disease and transgenic mouse models. Neuron. (1999) 24: 507-510.
13. Hutton M, et al. association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. (1998) 393: 702-705.
14. Weisgraber K, et al. The role of apolipoprotein E in the nervous system. Curr Opin Lipid. (1994) 5: 110-116.
15. Martorell L, et al. schizophrenic women with the APOE ´4 allele have a worse prognosis than those without it. Mol Psych. (2001) 6: 307-310.
16. Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. (1993) 261: 921-923.
17. Blacker D, et al. Apoe-4 and age at onset of Alzheimer’s disease. Neurology. (1997) 48: 139-147
18. Meyer M, et al. APOE genotype predicts when —not whether— one is predisposed to develop Alzheimer disease. Nat Genet. (1998) 19: 321-322.
19. Lambert JC, et al. Distortion of allelic expression of apolipoprotein E in Alzheimer’s disease. Hum Mol Genet. (1997) 6: 2151-2154.
20. Pericak-Vance M, et al. Complete genomic screen in late-onset familial Alzheimer disease. JAMA. (1997) 278: 1237-1241.
21. Hyman BT, et al. Role of the low-density lipoprotein receptor-related protein in b-amyloid metabolism and Alzheimer disease. Arch Neurol. (2000) 57: 646-650.
22. Pérez-Tur J. La genética y la enfermedad de Alzheimer. Rev Neurol. (2000) 30: 161-169.
23. Tanzi R. A genetic
dichotomy model for the inheritance of Alzheimer’s disease and common
age-related disorders. J. Clin. Invest. (1999) 104: 1175-1179.