Informaciones Psiquiátricas - Segundo trimestre 2007. Número 188
Envejecimiento y demencia en el Síndrome de Down*
Francisco Javier Ramón Jarne
Psiquiatra. Zaragoza.
Recepción: 26-02-07 / Aceptación: 02-04-07
INTRODUCCION
La población total que padece retraso mental en España, la constituyen aproximadamente 276.000 personas, de las que, entre 13.000 y 20.000 tienen una edad igual o superior a 65 años (Janicki, 1987).
El Síndrome de Down, es la causa no hereditaria más frecuente de retraso mental, y la segunda causa genética, después del Síndrome X frágil. De cada 1.000 recién nacidos uno tendrá Síndrome de Down, lo que supone el 16% de la población con retraso mental.
En la población con retraso mental, el grupo donde la esperanza de vida ha aumentado más en los últimos 20 años es el del Síndrome de Down, en el que, más de la mitad de los que superan los cinco años, superarán también los 40 años (JJ Eisering, 1987) y según estudios de Eyman et al. (1991) los institucionalizados, alcanzarán una esperanza de vida de más de 50 años.
El síndrome de Down (SD), fue descrito en el año 1866, por John Langdon Haydon Down. Posteriormente Jerôme Lejeune, en 1959 descubriría como causa del retraso mental la aparición de un cromosoma adscrito al número 21, el más pequeño de los cromosomas humanos.
Se considera que el SD está pues causado por la presencia de una tercera copia del cromosoma 21 (trisomía 21), existiendo un 2% de mosaicismo para la trisomía 21, que normalmente muestran menor afectación, y un 2% de traslocación de un segmento del cromosoma 21, a otro segmento, con frecuencia del cromosoma 14 (Dennis, 1999).
El aumento en la expresión de proteínas, por parte de los genes del cromosoma 21 da lugar a una cascada de efectos en el desarrollo de la estructura del cerebro fetal, que origina severas consecuencias tanto estructurales como funcionales que se manifiestan a lo largo de la vida de las personas con SD. Todavía no se conocen los mecanismos precisos que dirigen la aparición de los efectos específicos debidos a la dosis génica y su traducción en la conducta (Paterson, 1995). Existe una pequeña región en la parte distal del cromosoma 21 (21q22, 1-22.3), la región crítica SD, que está asociada a muchos de los rasgos del SD, en especial los faciales, la cardiopatía congénita, la estenosis duodenal, y algunos componentes del retraso mental. Existen otros «loci» fuera de esta región cromosómica, que parecen contribuir al fenotipo conductual completo, incluidas algunas formas leves de retraso mental, la aceleración del envejecimiento y la enfermedad de Alzheimer (Korenberg 1991; Epstein y col, 1992; Korenberg y col, 1994).
VARIABILIDAD DEL GENOTIPO Y CARACTERÍSTICAS FENOTÍPICAS CONDUCTUALES
El SD tiene unos rasgos típicos lo suficientemente específicos como para realizar el diagnóstico fácilmente en el periodo neonatal. Se han descrito hasta 300 rasgos diferentes, aunque ninguno es patognomónico, apareciendo en grado variable, siendo algunos edad dependientes. Algunos son sólo meros rasgos físicos sin repercusión fisiológica, otros tienen una importante gravedad y deben ser corregidos de inmediato, como las malformaciones cardíacas o las digestivas.
Los trastornos que se manifiestan en la edad adulta son las cataratas, la pérdida importante de agudeza auditiva, el hipotiroidismo y el prolapso de la válvula mitral. El retraso mental se asocia de manera constante al SD, los coeficientes intelectuales superiores a 70 son excepcionales, y la mayoría puntúa entre 35 y 50 en la escala de Wechsler. Parece no existir una correlación directa entre la expresividad fenotípica y la afectación mental.
La diversidad del fenotipo dependerá de la interacción entre los alelos de los genes, que se sobreexpresarán de modo diverso en los diferentes tejidos y en momentos distintos del desarrollo, y no de un único y defectuoso gen. La variabilidad en la expresión durante el desarrollo fetal puede afectar a la estructura del cerebro en formación, o ejercer su impacto en periodos críticos del desarrollo del cerebro, para producir una alteración selectiva en el funcionamiento cognitivo.
Los genes concretos cuya sobreexpresión tiene consecuencias graves, aunque no letales, sobre el desarrollo, pueden estar asociados a facetas diversas conductuales y fisiológicas del SD. Dos de estas facetas fisiológicas son el envejecimiento rápido, asociado a la superóxido dismutasa (SOD-1), y la enfermedad de Alzheimer, asociada al gen de la proteína precursora de beta-amiloide (APP), codificados en 21q21.1.
El desarrollo cognitivo en el SD se caracteriza, además de por alcanzar un CI promedio de 50, con oscilaciones entre 30 y 70, por un retraso adicional del lenguaje expresivo y de la memoria verbal a corto plazo (Chapman y col, 1999). La doble disociación de la memoria a corto plazo, verbal y visual es lo que caracteriza al SD en el que el procesamiento visual es superior al auditivo, con pobres habilidades lingüísticas, especialmente en la gramática, la articulación y el lenguaje expresivo en general, mientras que resulta significativa, la relativa habilidad en la memoria a corto plazo de naturaleza visual frente a la auditiva. Existe además una dificultad importante para la ejecución de los tests de repetición inversa de listados, tanto para secuencias verbales como visuales (Vicari y col, 1991), lo que revela la actuación de la función ejecutiva en la memoria operacional.
ENVEJECIMIENTO EN EL SÍNDROME DE DOWN
La esperanza media de vida de las personas con SD se aproxima a los 60 años y sin embargo continúa siendo inferior a la de la población general y a la de la población con deficiencia mental no debida al SD. Este síndrome es por tanto un claro factor de riesgo en lo que a la anticipación de la mortalidad se refiere (Strauss y Zigman, 1996), no obstante siguen apareciendo informes de personas con SD que alcanzan la década de los ochenta sin signos de demencia (Chicoine y McGuire, 1997).
Uno de los primeros trabajos que hablan de la asociación del SD y la EA es el de Jervis (1948). Un declive en las actividades de la vida diaria y en las habilidades cognitivas asociado a la edad, en una amplia muestra de individuos con SD , y que suponía un riesgo aumentado de signos clínicos de EA en torno a los 50 años de edad fue descrito en un estudio de Zigman & Schuff (1987).
Existe una clara relación entre síndrome de Down y enfermedad de Alzheimer (EA), esta relación se expresa en los siguientes hechos:
La tasa de aparición de EA en la población con SD es muy superior a la de la población general o a la de la población con deficiencia mental no debida a SD.
La edad de aparición de la EA en el SD es más temprana que en las poblaciones señaladas y es un factor que contribuye al acortamiento de la vida de las personas con SD.
Existen razones de naturaleza biológica que explican esta relación y en las que entraremos más adelante.
Es falsa sin embargo la opinión de que todas las personas con SD desarrollarán la EA cuando sean ancianas. Hay un declive natural y lento en el proceso de envejecimiento en las personas con SD sin demencia aunque éste pueda ser más precoz que en la población general, y un
declive con signos de demencia que define a la EA en el síndrome de Down., sin embargo existe una enorme variabilidad interindividual en la edad de comienzo de la demencia en las personas con SD y esto está relacionado con la prevalencia de la EA en el SD (tabla I).
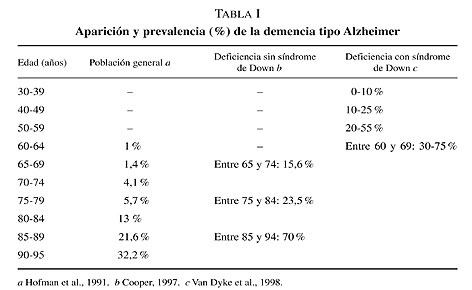
Las cifras expuestas indican claramente la mayor prevalencia de la EA en el SD, en comparación con otras poblaciones, y su tendencia a aumentar con la edad.
La edad a la que se presenta la EA en las personas con SD suele ser, como apuntábamos antes relativamente precoz, con una evolución media de 4-5 años.
La aparición y evolución muestran un alto grado de variabilidad interindividual, y hay personas con SD que nunca desarrollarán demencia.
Para que aparezca la EA deben pues confluir factores diversos cuya expresión difiere de una persona a otra.
Hay que distinguir entre lo que es un deterioro o declive asociado a la edad en el SD y lo que es la instauración de un proceso demencial. Por eso es necesario hacer un buen diagnóstico diferencial entre un cuadro de demencia y otros procesos que pueden aparecer en el SD y que cursan con deterioro cognitivo como los trastornos depresivos, el hipotiroidismo y las pérdidas sensoriales.
El diagnóstico de demencia requiere el conocimiento del nivel de funcionamiento previo tanto en los aspectos cognitivos como en los aspectos funcionales de habilidades y conductas adaptativas.
Se deben considerar como síntomas prodrómicos las pérdidas en las habilidades de la memoria, el lenguaje, la comunicación, y la orientación general. Aparecerán posteriormente cambios de personalidad, periodos crecientes de apatía e inactividad, hiperreflexia, crisis convulsivas, pérdidas de las habilidades adaptativas, de la memoria visual, reducción del lenguaje expresivo, confusión y desorientación crecientes, y aumento de las estereotipias.
El primer riesgo es pues considerar que cualquier cambio o deterioro cognitivo o de carácter conductual adaptativo en un adulto con SD, significa el inicio de una demencia tipo Alzheimer (tabla II).

Es necesario considerar un conjunto de cuadros patológicos que pueden aparecer en personas con síndrome de Down y que cursan con cuadros sindrómicos que pueden parecerse a una demencia.
Es necesario tener en cuenta que según McGuire y Chicoine (1997) un porcentaje importante de personas con SD han podido ser diagnosticadas de demencia cuando tenían realmente otros cuadros patológicos que podían haber sido tratados con éxito, al ser potencialmente reversibles. Con frecuencia pueden iniciarse cuadros depresivos coincidiendo con acontecimientos de toda índole que ocurren en la vida del adulto con SD, y que por dificultades de expresión y procesamiento pobre de la información o falta de estrategias de afrontamiento pueden cursar con trastornos de conducta.
Otras veces son las pérdidas sensoriales, visuales o auditivas o la aparición de hipotiroidismo, que si no son reconocidas o expresadas por el paciente pueden ocasionar graves modificaciones de conducta. Así en el paciente adulto SD que presenta síntomas compatibles con un proceso de demencia es necesario hacer un buen screening que descarte otras posibilidades diagnósticas. Será necesario analizar la función tiroidea, los niveles de ácido fólico, y vitamina B12, la fórmula sanguínea, la velocidad de sedimentación y la presencia de autoanticuerpos para descartar anemias, infecciones y enfermedades autoinmunes, así como realizar exploraciones neurológicas completas con técnicas complementarias de EEG y resonancia magnética. Visser y col, 1996 han destacado la importancia del EEG como herramienta diagnóstica de la demencia en las personas con SD ya que han descrito un enlentecimiento progresivo del ritmo occipital dominante que guarda relación con el deterioro cognitivo.
Es necesaria una evaluación cognitiva y conductual, mediante el uso de escalas psicométricas adaptadas a la población con deficiencia mental y específicamente a la población con SD, que partiendo de un conocimiento del nivel previo nos permitan valorar el deterioro (tabla III).

De los estudios realizados hasta ahora hay que destacar el realizado por Lai & Wiliams (1989) de carácter prospectivo para evaluar la prevalencia de EA en 96 pacientes con SD por encima de los 35 años, de los que 49 cumplían criterios de demencia. La prevalencia de EA fue del 8% en el grupo de 35-49 años, del 55% en el de 50-59 años y 75% por encima de los 60 años.
También es importante el de Visser y col (1997), de tipo longitudinal, en 307 sujetos y que abarcó un periodo de hasta 10 años de evolución. De los sujetos de la muestra 56 desarrollaron demencia, siendo la media de edad de inicio de 56 6 97,4 años (intervalo entre 40,9 y 72 años), estableciendo como criterios diagnósticos de inicio de demencia la aparición de al menos 10 síntomas incluidos en su Early Signs of Dementia Checklist, la pérdida de al menos un 25% de habilidades en el Inventario de Habilidades sociales para personas con retraso mental, y la reducción de al menos 1 Hz en el ritmo dominante de base del EEG en la región occipital. Había personas que también mostraban deterioro, pero en grado menor y presentaban además modificaciones de conducta, pero no podían ser diagnosticadas de demencia. Este estudio constataba que había sujetos con SD que desarrollaban demencia y otros que mostraban cierto declive o deterioro en su evolución relacionado con la edad sin que por ello presentaran demencia. Se concluía que existía un declive funcional en los ancianos con SD, que era propio de la edad y no de la enfermedad de Alzheimer. El deterioro descrito en el estudio se caracterizaba por síntomas iniciales poco específicos con disminución del interés, de la dificultad con que se mueven, de la motivación, cambios de humor o exageración de sus rasgos psicológicos. Progresivamente disminución de la capacidad de realizar movimientos coordinados, y de ejecutar las tareas habituales, descuido en el cuidado personal y disminución de la capacidad de comunicarse verbalmente, posteriormente pérdida progresiva de la orientación temporoespacial y dificultad para la deambulación, y por último, incontinencia esfinteriana y crisis comiciales.
Prasher y col. (1998) en otro estudio longitudinal y prospectivo realizado en 128 sujetos con SD a lo largo de 3 años insiste en que un deterioro asociado a la edad en las capacidades adaptativas no significa que haya una evolución hacia la enfermedad de Alzheimer. Para el diagnóstico de demencia exigen la evidencia de importantes cambios en la cognición, el humor, la conducta y el funcionamiento social, de acuerdo con los criterios diagnósticos de la DCR-10 (Diagnostic Criteria for Research) establecido por la OMS en 1993, y que se mantengan o agraven durante un mínimo de 2 años. Prasher establece dos grupos, el de los que presentan demencia y el de los que presentan deterioro sin demencia.
En los que no presentan demencia los síntomas relacionados con el funcionamiento independiente, puntuaron significativamente más altos a lo largo de los 3 años que en el grupo que presentaba demencia, y a la inversa ocurría en los síntomas relacionados con la inadaptación. En el grupo sin demencia el deterioro fue menor o inexistente, cuando no existía ninguna otra patología asociada.
Devenny y col (1997) en otro estudio longitudinal de 91 sujetos entre 50 y 63 años, a lo largo de 6 años concluye que con la excepción de 4 casos, el resto de sujetos mantenía esencialmente invariables sus niveles iniciales de funcionamiento. Estos autores proponen la necesidad de distinguir el envejecimiento normal que se inicia precozmente en el SD con respecto a la población general, aunque el deterioro progresa lentamente; de la evolución hacia la demencia. Algunos aspectos de los resultados concuerdan con la sugerencia de que los adultos con SD pueden experimentar un envejecimiento prematuro pero por lo demás normal. Entre otras conclusiones de este estudio, cabe citar, la disparidad entre los hallazgos clínicos y los neuropatológicos relacionados con la EA, que en los adultos con SD parece ser un fenómeno real y que el riesgo de EA en los adultos con SD que poseen un nivel alto de funcionamiento y entran en su sexta década es considerablemente inferior al indicado por estudios anteriores.
En todos los estudios destaca una importante variabilidad interindividual en la edad de comienzo de la demencia en las personas con síndrome de Down y ello está estrechamente relacionado con la prevalencia de la enfermedad de Alzheimer en esta patología. Modificaciones en el área cognitiva que se detectan con relativa facilidad en la población general son más difícilmente detectables en las personas con deficiencia mental, por ello es necesario analizar con mayor precisión las modificaciones de carácter adaptativo y conductual y distinguir las que son propias de un envejecimiento natural de las que son propias de un envejecimiento patológico. Aunque puede admitirse la existencia de una mayor prevalencia de la enfermedad de Alzheimer en el SD, en comparación con otras poblaciones y su importante tendencia a aumentar con la edad, hay que destacar los amplios intervalos en las cifras referidas al SD que tienen los datos de los estudios de Haveman y col (1995), Schupf y col (1989), Lai y Williams (1989), Zigman y col (1995), Prasher (1995) y Visser y col (1997).
LA NEUROPATOLOGÍA TIPO ALZHEIMER EN EL SÍNDROME DE DOWN
La presencia de neuropatología tipo Alzheimer en los cerebros de sujetos con SD es una constante que fue establecida de forma definitiva hace ya dieciocho años. Se encuentran hallazgos neuropatológicos similares a los de la EA, en casi todos los pacientes con SD mayores de 40 años, mientras que la constatación de deterioro cognitivo en adultos con SD es muy variable en la literatura. En el SD se ha demostrado un descenso en la densidad sináptica de la corteza sensitivo motora entre las semanas 32 y 34 de la gestación, así como una reducción de las hendiduras pre y postsinápticas. Existe una anomalía en el desarrollo dendrítico y una alteración en la expresión proteica. Estas alteraciones afectarían a la transmisión sináptica que podría ser la causante de las alteraciones cognitivas y motoras de la enfermedad. La preexistencia de estas anomalías congénitas unidas a las lesiones neuropatológicas similares a las de la EA, podría facilitar la aparición de un deterioro en las edades medias de la vida de las personas con SD. Por otra parte el envejecimiento ocurre a edades tempranas en el SD y ello actuaría como factor facilitador. Estudios del cerebro con RM, demuestran que en el SD se encuentran frecuentemente tres marcadores de envejecimiento: atrofia cortical, lesiones en la sustancia blanca, e hipointensidad de los ganglios basales en T2. Los estudios de flujo cerebral y de utilización regional de la glucosa se afectan a partir de los 45 años. Antes de que se desarrolle la EA, en el SD, conforme avanza la edad disminuyen los volúmenes de la amígdala, el hipocampo, y la circunvolución parahipocámpica posterior de forma simétrica, y existe una correlación positiva entre estas alteraciones del lóbulo temporal medial y distintos pruebas de memoria en adultos con SD, que no han desarrollado demencia (Krasuski y col, 2002).
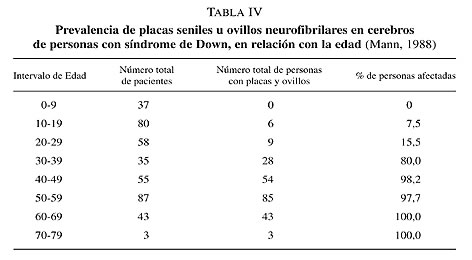
Desde el punto de vista estructural, el cerebro es de bajo peso y la atrofia es de distribución global, afectando especialmente al lóbulo temporal medial y la corteza asociativa de los lóbulos temporales, parietales y frontales, mientras que la corteza primaria y las estructuras subcorticales, están más preservadas. En la segunda o tercera décadas, es posible detectar la presencia de depósitos de Ab. Una década después aparecen las placas neuríticas y los ovillos neurofibrilares. La distribución de las lesiones y su neuroquímica es similar a la descrita en la EA. La existencia de algunas diferencias morfológicas en las placas neuríticas, podría deberse al curso más prolongado de la neuropatología de la demencia en el SD que en la EA, como el hecho de que las placas sean más grandes en el SD que en la EA (tabla IV).
Esta neuropatología viene definida tanto en el SD como en la EA por la presencia de lesiones cerebrales específicas:
Los depósitos de la proteína b-amiloide que originan las placas de localización extraneuronal.
La acumulación de detritos neurofibrilares intraneuronales que constituyen los ovillos o acúmulos neurofibrilares de localización intraneuronal.
Pérdida de neuronas y de sus uniones sinápticas.
Otras alteraciones como son las modificaciones vacuolares de las neuronas y de los cuerpos de Lewy y de Hirano.
Hay una distribución selectiva y jerarquizada de las lesiones neurofibrilares y en menor medida, de las placas seniles, que son menos selectivas en su distribución que se produce con mayor uniformidad entre las áreas asociativas. Esta disociación de la distribución de las lesiones más características de la EA, evidencia la cuestión no resuelta de la relación entre ambas.
En los estadíos I y II de Braak y Braak los acúmulos neurofibrilares se localizan en la corteza transentorrinal, entorrinal y subcampos CA1/subiculares del hipocampo. En los III y IV, hay daño intenso de la corteza entorrinal y perirrinal, moderada en CA1/subículum y leve en CA4, y ya hay afectación de amígdala, núcleos basales, porciones límbicas del tálamo y en pequeña escala áreas corticales asociativas. En los V y VI, presencia de afectación neurofibrilar en casi todas las áreas asociativas, que se extiende a múltiples regiones de toda la corteza cerebral con pérdida escalonada y progresiva de neuronas de diverso carácter neuroquímico, siendo frecuente que las primeras neuronas afectadas sean colinérgicas. La corteza termina por atrofiarse y se dilatan notablemente los ventrículos cerebrales.
BASES NEUROBIOLÓGICAS DE LA RELACIÓN ENTRE SÍNDROME DE DOWN Y ENFERMEDAD DE ALZHEIMER
La asociación de demencia y SD se conoce desde los trabajos de Jervis en 1948. El primer gen relacionado con la enfermedad de Alzheimer en 1987 (Mann DM, Esiri MM) es el gen de la proteína precursora del amiloide, localizado en el brazo largo del cromosoma 21, dadas las similitudes de las lesiones que aparecían en los sujetos con SD con la edad. Las mutaciones de este gen en la población general dan lugar a la síntesis de un amiloide anormal que genera amiloidosis cerebral, afectando a varios grupos familiares con una penetrancia dominante y una edad de inicio media de 50 años.
El amiloide beta (Ab) es un producto del metabolismo de la PPAb codificada por el gen localizado en el brazo largo del cromosoma 21. La PPA es una glucoproteína de membrana y su función fisiológica, parece estar relacionada con mecanismos de transmisión o adhesión celular en las membranas celulares, y se expresa tanto en el SNC (en neuronas, microglía y astrocitos), como en órganos periféricos. La PPA tiene una secuencia de 770 aminoácidos como máximo, y presenta varias isoformas, de las que se expresan con mayor frecuencia en el SNC las PPAb770, 751, 714 y especialmente, la 695, precisamente, las más amiloidogènicas. El procesamiento metabólico de la PPA conlleva una serie de roturas proteolíticas que liberan el Ab de 39-43 aminoácidos. Dos de los tercios del péptido 28 aminoácidos pertenecen a la región extracelular del PPA, y el tercio restante está contenido dentro de la membrana celular.
Existen como mínimo dos vías para el metabolismo de la PPA, que implican tres cortes en su secuencia debido a la intervención de varias enzimas secretasas (abg) lo que implica la producción de distintos fragmentos.
La secretasa a actúa en la superficie celular y rompe la PPA entre los aminoácidos 16 y 17 del Ab, y, por lo tanto no es amiloidogénica. Produce una fracción larga llamada Nexina 2, soluble, que se expulsa hacia el espacio extracelular y es inocua, incluso se piensa que puede tener un efecto neurotrófico. El aumento de la actividad a-secretasa podría frenar la vía amiloidogénica y ello podría utilizarse con finalidad terapeútica, entre las candidatas al papel de a-secretasa la familia de las metaloproteínas denominadas ADAM, son objeto de estudio como dianas terapéuticas.
Existe otra vía, localizada en el compartimento lisosómico, con la intervención sucesiva de dos enzimas, una b-secretasa y posteriormente una g secretasa, que liberan un péptido b-amiloide de cadena corta, el cual puede estar en tres formas a su vez: Ab40, Ab42, y Ab43. La forma corta Ab40 es la predominante en el metabolismo normal (el Ab se encuentra presente en forma soluble en el plasma y en el LCR de sujetos no demenciados), mientras que los péptidos Ab42 y 43 tienen una gran capacidad neurotóxica y fibrilogenética, por lo que precisamente la agregación de cúmulos de estas fracciones es el inicio de la formación de las placas amiloideas.
En 1999 se caracterizó un gen situado en el cromosoma 11 que codifica una enzima aspartato proteasa transmembrana humana, ampliamente difundida en tejido cerebral, denominada BACE1, cuyas características coinciden con las de la b-secretasa, codifica una proteína de 501 aminoácidos, con un único potencial segmento transmembrana. El sitio activo estaría en la luz vesicular del aparato de Golgi y de los endosomas. Existe también otra enzima homóloga, la BACE2, más difundida en el tejido periférico y codificada por un gen situado en el cromosoma 21 y que parece no tener influencia en el desarrollo de la enfermedad de Alzheimer. Tras la b-secretasa, actuaría la g-secretasa, efectuando un corte no muy preciso en la cercanía del residuo 712 de la PPA. Aunque hasta ahora no se ha determinado la composición de la g-secretasa se cree que se trataría de una estructura macromolecular compleja, de la que formarían parte las presenilinas y otras proteínas como la nicastrina.
Se ha evidenciado que el amiloide se deposita en forma de agregados de fibras de polímeros con estructura de lámina beta plegada, en el espacio extracelular, formando las placas seniles o la angiopatía congófila vascular encontrada en la enfermedad de Alzheimer. Las placas seniles son más habituales en el hipocampo y a nivel parietal, mientras que la amiloidosis vascular lo es en la zona occipital.
La hipótesis de la «cascada amiloide» afirma que la sobreproducción del péptido Ab es el hecho básico en la etiología de la enfermedad. El depósito de la Ab fibrilar es neurotóxico para la formaciones nerviosas y desencadena allí donde se acumula, la respuesta neuronal que concluye en la formación de ONF (ovillos neurofibrilares) y en la muerte celular, a través de mecanismos que pueden incluir una alteración de la respuesta inmune/inflamatoria, la disregulación de los canales de calcio (elevación de los niveles de calcio intracelulares), o la sobreproducción de elementos oxidantes y radiales libres, lo que puede llegar a producir la muerte celular por apoptosis. Cuando se inicia el envejecimiento cerebral aumenta notablemente la síntesis de este péptido. En la enfermedad de Alzheimer, la cantidad de amiloide es notablemente superior y predomina la forma Ab42.
La presenilina 1 codificada en un gen del cromosoma 14, interviene en la enfermedad de Alzheimer, favoreciendo la síntesis de amiloide b. Se postula que las mutaciones que ocurren sobre este gen incrementan esa función. La presenilina 2 codificada por un gen en el cromosoma 1 tiene una actividad similar aumentando la producción de Ab.
La apoE es una proteína plasmática implicada en el transporte de colesterol y otros lípidos en los diferentes tejidos, codificada por un gen situado en el cromosoma 19. Presenta un polimorfismo en su expresión, con tres isoformas principales E2. E3 y E4 (codificadas por los alelos e2, e3 y e4 respectivamente). La relación entre la apoE y la enfermedad de Alzheimer, se establece por la representación exagerada de la forma E4 en los sujetos afectados por la enfermedad. Existen indicios del mecanismo patogénico conectado con el alelo e4, relacionándolo con un aumento del depósito de amiloide dado que se han aislado fragmentos de apoE en las placas seniles. La forma E4 presenta una capacidad de unión al péptido beta, muy superior a la de E3, también puede facilitar la hiperfosforilación de la proteína tau y favorecer la creación de ONF.
La degeneración neurofibrilar en la enfermedad de Alzheimer se manifiesta por la presencia de ovillos neurofibrilares (ONF), de los hilillos neurofibrilares, y de las placas neuríticas. Todas estas estructuras, están formadas por acumulaciones de filamentos helicoidales apareados, cuyo elemento fundamental es la proteína tau hiperfosforilada y poco soluble. La proteína tau normal es soluble, está asociada a los microtúbulos axonales y desempeña un papel importante en la polimerización y estabilización de los mismos. Se sintetiza a partir de un único gen situado en el cromosoma 17 y tiene seis isoformas. En la enfermedad de Alzheimer, la proteína tau está hiperfosforilada e hiperglucoxilada, y presenta un patrón de fosforilación distinto al de la proteína normal y la consecuencia es una disminución de su capacidad de unión a los microtúbulos, por lo que éstos se despolimerizan, alteran el transporte axonal y la neurotransmisión sináptica, lo que provoca la muerte neuronal. La hiperfosforilación de tau, probablemente se debe a una hiperactivación de las cinasas o a una desactivación de las fosfatasas, en este sentido el péptido Ab puede favorecer la hiperactivación de la taucinasa tipo II.
Partiendo de la hipótesis de la cascada amiloide y puesto que la proteína APP es codificada por un gen del cromosoma 21, parece lógico que si existe una trisomía de este cromosoma, haya una sobreexpresión de la proteína APP, dato confirmado desde edades precoces en sujetos con síndrome de Down.
En el síndrome de Down, se aprecia un aumento muy precoz en la formación de APP, con sustanciales incrementos tanto en el plasma sanguíneo como en el cerebro de los sujetos afectados por este síndrome, a edades muy tempranas, y muy anteriores que en el resto de la población general Sin embargo no basta sólo con que haya un exceso de producción de APP, sino que su procesamiento dé lugar a Ab42, que es lo que ocurre en el Síndrome de Down. De hecho es posible observar aumentos de los niveles plasmáticos de Ab42 en los sujetos con el síndrome de Down (Schupf y col, 2001), así como Ab42 soluble en los cerebros de estos sujetos, en edades muy precoces, incluso prenatales (Teller y col, 1996; Lemere y col, 1996; Mori y col, 2002). Este Ab42 tan precozmente detectable, se aprecia primero localizado intracelularmente, y posteriormente pasa al espacio etracelular y se agrega a las placas amiloides.
El mecanismo por el cual en el síndrome de Down, la APP producida en exceso de lugar a la formación de Ab42 neurotóxica, es la gran cuestión que se está intentando resolver.
ESTRÉS OXIDATIVO Y FORMACIÓN DE Ab42
Se piensa que el factor responsable de la activación protagonizada por la acción de la b-secretasa sea el estrés oxidativo en las mitocondrias neuronales, provocado por la acumulación intracelular de especies reactivas al oxígeno (ROS). La mayoría de los radicales libres presentes en el organismo (elementos que pueden existir independientemente y que poseen uno o más electrones no emparejados) son especies reactivas al oxígeno, como aniones superóxidos, radicales hidróxilos, agua oxígenada, etc. Estos radicales son muy reactivos, y tienen gran capacidad para dañar macromoléculas celulares, incluidos los ácidos nucleicos. Las mitocondrias son fuente importante de ROS, pues es el lugar donde se consume la mayor parte del oxígeno usado en la célula, además de poseer en el metabolismo normal, la mayor capacidad de neutralización de esas sustancias. Durante el envejecimiento se produce una disminución del funcionamiento mitocondrial y este órgano es muy susceptible al daño oxidativo. Sin embargo ya en niños con SD se detectan niveles significativamente elevados de carbonil-proteínas, en comparación con controles sanos, mientras que los niveles de 4-hidroxi-2-nonenal y la capacidad antioxidante era similar en ambos grupos, lo que confirmaba un incremento de estrés oxidativo en pacientes con SD responsable de daño oxidativo a proteínas (Zitnanova I y col. 2006).
Busciglio y col 2002 en cultivos de células obtenidos de cerebros fetales con síndrome de Down obtiene los siguientes resultados:
En corteza cerebral, en cultivo de astrocitos, El nivel de APP está aumentado en relación con los no SD. El nivel de C83 (que depende de la actividad de la a-secretasa) está disminuido., mientras que el de C99 (que depende de la b-secretasa) está aumentado. Los astrocitos muestran una disminución de la secreción de APP y Ab, con acumulación intracelular de Ab en forma de agregados insolubles dentro de compartimentos subcelulares asociado con la circulación y el procesamiento de APP.
Los cultivos de neuronas, muestran antes del 5º día de cultivo el aumento de la presencia de APP, acumulación intracelular de Ab, con disminución de la secreción de Ab y APP.
La depleción energética provocada por un veneno mitocondrial en los astrocitos no SD, incrementó la presencia intracelular de APP y C99, mientras que redujo la concentración de C83 y APPs soluble, de forma similar a los cambios observados en los astrocitos SD, detectándose la presencia de agregados Ab42 en todo el citoplasma.
Existe una disminución del potencial transmenbrana en las mitocondrias de los astrocitos SD, con disminución de la actividad redox.
En cerebros humanos de sujetos con SD, existe, tanto en neuronas como en astrocitos, una acumulación intracelular de Ab42, que puede preceder a la formación de placas amiloides y acúmulos neurofibrilares, en cambio existe una reducción de APP soluble. En cultivos de neuronas corticales SD la incorporación de APP soluble impidió la neurodegeneración, y esto no ocurrió en neuronas normales, lo que sugiere que posee un valor neuroprotector.
PAPEL DE LAS PRESENILINAS Y LA apoE4
Además del estrés oxidativo, en la formación de Ab42, deben intervenir otras proteínas transmenbrana como la PS-1, la PS-2, y la proteína apoE4. La presencia del alelo e4 es un factor de riesgo en el desarrollo de la enfermedad de Alzheimer, (Polvikoski y col. 1995), y se ha observado que contribuye a su instauración en las personas con Síndrome de Down (Schupf y col. 1996), apareciendo el alelo e2 como un factor protector (Prasher y col. 1997). En su estudio en sujetos con síndrome de Down, Prasher, no llegó a evidenciar una asociación significativa entre la posesión del alelo e4 y el inicio de la enfermedad de Alzheimer, si bien los sujetos con este alelo mostraron una mayor tendencia a que la enfermedad se iniciara a edades más precoces, mientras que las personas con el alelo e2 podían no desarrollar la enfermedad y tener una longevidad mayor.
La presencia del alelo e4 influye en el procesamiento de Ab, pudiendo incrementar la precipitación y el acúmulo de Ab43 y Ab42 (McNamara y col, 1998). De hecho, Schupf y col, en 2001 evidenciaron que los niveles plasmáticos de Ab42 estaban más aumentados en los adultos con síndrome de Down que mostraban signos de demencia que en los que no los mostraban, y que la presencia del alelo e4 estaba relacionada con una elevación de los niveles de Ab42 y no con los de Ab40.
Se desconoce todavía el mecanismo por el que el alelo e4 de la apoE, facilita la mayor presencia y deposición de Ab42 ; si se trata de una mayor velocidad en la formación de fibrillas o de una menor velocidad en la limpieza y desaparición de las mismas. Lo más interesante no obstante, es la demostración en cultivos de células musculares lisas de que la presencia concurrente de estrés oxidativo, favorece la acumulación de apoE4 y la formación de depósitos apoE4-Ab; y estos depósitos a su vez, favorecen la aparición de una mayor peroxidación lipídica (Mazur-Kolecka y col, 2003).
Es posible que el trastorno en el metabolismo energético de las células síndrome Down, consecuencia del estrés oxidativo al que se ven sometidas, sea un factor que contribuya a promover la actividad de la b-secretasa, y de este modo facilite la ruptura de la APP en el lugar inadecuado, alterando además la circulación de Ab42 de manera que se acumule intracelularmente formando agregados insolubles que ejercen una acción neurotóxica.
También es posible que esta acumulación intracelular, tan precozmente iniciada, tenga que ver con otro fenómeno, descrito recientemente, la presencia intracelular de grandes desmosomas. Estas estructuras son ricas en proteasas incluida la b-secretasa, e intervienen en los procesos de intercambio, reciclaje y modulación catabólica de diversas macromoléculas. Se ha comprobado la presencia de desmosomas anómalos en las primeras etapas de la enfermedad de Alzheimer, su actividad contribuye al procesamiento anormal de la APP por parte de la b-secretasa, y se ha asociado de hecho la presencia de estos desmosomas con la producción excesiva de Ab.
Se ha demostrado que el mismo tipo de desmosomas anómalos se encuentra muy precozmente en las neuronas del síndrome de Down (Cataldo y col, 2000), incluso antes del nacimiento, en estructuras cerebrales como el hipocampo, la corteza y los ganglios de la base. Es posible que la propia sobreexpresión de APP en el síndrome de Down sea uno de los factores que alteren la función mitocondrial, o que el mismo Ab42 por su acción tóxica altere la función mitocondrial, o que el exceso de actividad necesaria para aclarar o limpiar los agregados de proteínas, suponga un fuerte coste metabólico para la célula, debido a la gran cantidad de energía que requiere, y eso favorezca la alteración mitocondrial. La existencia de un alelo e4 constituye un factor adicional, no determinante, pero que contribuye a la iniciación de la amiloidogénesis patológica; además el propio estrés oxidativo puede facilitar la acción patogénica de la apoE4.
Hay que señalar que existe una actividad de fondo caracterizada por la presencia de estrés oxidativo en las células de los organismos con SD, y su intensidad, puede ser muy variable, dependiendo del grado en que se sobreexprese el gen de la SOD1, presente en el cromosoma 21, así como de otros factores, como el exceso de APP. Esta acción es permanente, y su influencia se expresa sobre las células de distintos tejidos a lo largo de la vida, a las que somete a un desgaste constante que puede explicar la precocidad con que aparece el envejecimiento en el síndrome de Down. Al mismo tiempo, la lesión de las estructuras mitocondriales, inherente al estrés oxidativo, desencadena la activación de la b-secretasa, junto con otros factores coadyuvantes. Y sobre el fondo de una sobreproducción de APP debida a la sobreexpresión del gen presente en el cromosoma 21, se dan las condiciones para ir produciendo a lo largo de los años el exceso de Ab42 neurotóxico. La intensidad con la que esta proteína se produce, la velocidad con la que se genera, y la distribución por los núcleos y áreas cerebrales, van a ser los condicionantes de la aparición de la enfermedad de Alzheimer, de su precocidad, de la intensidad de sus síntomas, y de la rapidez de su evolución.
TERAPIAS FARMACOLÓGICAS
En una revisión de Ezio Giacobini (Pharmacological Research 50; 2004; 433-440) se analiza la respuesta al donepezilo, uno de los inhibidores de la acetilcolinesterasa, en el tratamiento de un pequeño número de pacientes con síndrome de Down y enfermedad de Alzheimer en un periodo de 8 a 40 semanas. Se describen efectos clínicos de disminución de la confusión y mejora del funcionamiento cognitivo. En otro estudio realizado por Boada-Rovira M y col. en 2005, se informa, que donepezilo parece ser efectivo en el tratamiento de las alteraciones cognitivas y conductuales asociadas a la EA en el SD, pero que hasta ahora las muestras utilizadas son pequeñas y los periodos de exposición al tratamiento son cortos para tener resultados más concluyentes. Prasher VP y col en 2004, afirma que tanto, donepezilo como rivastigmina, galantamina o memantina, pueden jugar un papel importante en el manejo de pacientes con EA y SD, y ello parece razonable dadas las similitudes neuropatológicas y neuroquímicas entre ambos trastornos (tabla V).

Dado que en muchos aspectos el síndrome de Down puede ser un modelo preclínico de la enfermedad de Alzheimer, es necesario valorar el uso precoz de los inhibidores de la acetilcolinesterasa y otros fármacos antioxidantes, hipocolesterolémicos, o inhibidores de la producción del péptido betamiloide, con finalidades no sólo terapeúticas sino también preventivas.
BIBLIOGRAFÍA
1. Aylward Eh, Burt Db, Thorpe Lu, Lai F, Dalton A. Diagnosis Of Dementia In Individuals With Intellectual Disability. Jintellec Disabil Res 1997; 42: 152-164,1997.
2. Becker L Et Al. Growth And Development Of The Brain In Down Syndrome.» The Morphogenesis Of Down Syndrome». Wiley-Coss Eds. 1991. Pp 133-152.
3. Burt Db, Loveland, Ka, Lewis Kr. Depression And The Onset Of Dementia In Adults With Mental Retardation. J Merit Retard 1992; 96: 505-522.
4. Carlesimo G, Marotta L, Vicari S. Long-Term Memory In Mental Retardation: Evidence For A Specific Impairment In Subjects With Downs Syndrome. Neuropsychologia 1997; 35: 71-79.
5. Caselli Mc, Vicari S, Longobardi E, Et Al. Gestures And Words In Early Development Of Children With Down Syndrome. J Speech Lang Hear Res 1998; 41: 1125-1135.
6. Cataldo Am, Peterhoff Cm, Troncoso Jc, Gómez-Isla T, Hyman Bt, Nixon Ra. Endocytic Pathway Abnormalities Precede Amyloid Beta Deposition In Sporadic Alzheimers Disease And Down Syndrome: Differential Effects Of Apoe Genotype And Presenilin Mutations. Am J Pathol 2000; 157: 277-286.
7. Chapman Rs. Language And Cognitive Development In Children And Adolescents With Down Syndrome. En: Miller Jf, Leavitt La, Leddy M, Eds. Improving The Communication Of People With Down Syndrome. Baltimore: Brookes 1999, P 41-60.
8. Chicoine B, Mcguire D. Longevity Of A Woman With Down Syndrome: A Case Study. Merit Retard 1997; 35: 477-479.
9. Cooper S-A, Prasher Vp. Maladaptive Behaviours And Symptoms Of Dementia In Adults With Downs Syndrome Compared With Adults With Intelectual Disability Of Other Aetiologies. J Intellect Disab Res 1998; 42: 293-300.
10. Cooper Sa. High Prevalence Of Dementia Amongst People With Learning Disabilities Not Attributed To Down Syndrome. Psychol Medic 1997; 27: 609-616.
11. Dennis J. Psychological And Behavioral Phenotypes In Genetically Determined Syndromes: A Review Of Research Findings: Down Syndrome. En: Obrien G, Yule W, Eds. Behavioral Phenotypes. London: Mac Keith Press. 1995 P. 105-109.
12. Devenny Da, Silverman Wp, Hill Al, Jenkins E, Sersen Ea, Wisniewski Kw, Envejecimiento Normal En Adultos Con Síndrome De Down: Un Estudio Longitudinal. Rev Síndrome Down 1997; 14: 94-104.
13. Devenny, D.A.Et Al (1992) Ageing In Higher Functioning Adults With Downs Syndrome: An Interim Report In A Longitudinal Study. Journal Of Intelectualdisability Research,Vohxmen 36, 241-250.
14. Down Jl. Observation On An Ethnic Classification Of Idiots. London Hospital. Clinical Lectures And Reports 1866; 3: 259-262.
15. Eisering, JJ (1987). Aspectos Del Envejecimiento En El Síndrome De Down Sigh Cero, 113,42-48.
16. Epstein Cj, Korenberg J, Anneren G, Et Al. Protocols To Establish Genotype-Phenotype Correlations In Down Syndrome. Am J Hum Genet 1991; 49: 207-235.
17. Evenhuis Hm. Evaluation Of A Screening Instrument For Dementia In Aging Mentally. Retarded Persons. J Intellect Disabil Res 1992; 36: 337-347.
18. Evenhuis Hm. Medical Aspects Of Ageing In A Population With Intellectual Disability: Iii. Mobility, Internal Conditions And Cancer. J Intel Disabil Res 1997; 41: 8-18.
19. Evenhuis Hm. The Natural History Of Dementia In Ageing People With Intellectual Disability. J Intel Disabil Res 1997; 41: 92-96.
20. Evenhuis Hm. The Natural History Of Dementia In Downs Syndrome. Arch Neurol 1990; 47: 263-267.
21. Eyman, R Et Al (1991) Life Expectancy Of Persons With Down Syndrome. American Journal On Mental Retardation, Vol 95, N.o 6, 603-12.
22. Florez J. Aspectos Médicos Del Anciano Con Deficiencia Mental. En: Gafo J (Ed). Deficiencia Mental Y Problemas Éticos En Torno Al Final De La Vida Madrid, Pub. Univ. Pontificia Comillas 2000.
23. Florez J. Envejecimiento Y Síndrome De Down. ¿Alzheimer, Si O No? Rev Síndrome Down 1993; 10: 55-62.
24. Florez J. Nuevos Tratamientos. Rev Síndrome Down 1999; 16: 49-51.
25. Florez J. Terapéutica Farmacológica De Las Demencias. Medicine 1998; 7(99): 4633-4643.
26. Florez, J (1993) El Envejecimiento De Las Personas Con Síndrome De Down: ¿Alzheimer, Si O No? Revista Síndrome De Down Cantabria. Set. 55-62.
27. Gedye A. Dementia Scale For Down Syndrome. P.O. Box 39081. Point Grey, Vancouver.
28. Gibson Et Al (1988) Age And Pattern Of Intelectual Decline Among Downs Syndrome And Other Mentally Retarded. International Journal Of Rehabilitation Research, Vol 11, 47-55.
29. Hofman A, Rocca Wa, Brayne C, Et Al. The Prevalence Of Dementia In Europe: A Collaborative Study Of 1989-90 Findings. Int Jepidemiol 1991; 20: 736-748.
30. Hyman Bt, West Hl, Rebeck Gw, Buldyrev Sv, Mantegna Rn, Ukleja M, Havlin S, Stanley He. Quantitative Analysis Of Sennile Plaques In Alzheimer Disease: Observation Of Log-Normal Size Distribution And Molecular Epidemiology Of Differences Associated With Apolipoprotein E Genotype And Trisomy 21 (Down Syndrome). Proc Natl Acad Sci 1995; 92: 3586-3590.
31. Janicki, M (1987). Perspectiva General Del Envejecimiento Y La Deficiencia Mental. Siis-Real Patronato De Pre. Y Atención A Personas Con Minusvalía. Colección Documentos, Pag. 37.
32. Jervis Ga. Early Senile Dementia In Mongoloid Idiocy. Am J Psychiatry 1948; 105: 102-106.
33. Johannsen P, Christensen Je, Mai J. The Prevalence Of Dementia In Down Syndrome. Dementia 1996; 7: 221-225.
34. Kishnani Ps, Sullivan Ja, Walter Bk, Spiridigliozzi Ga, Doraiswamy Pm, Krishnan Krr. Cholinergic Therapy For Downs Syndrome. Lancet 1999; 353: 1063-1065.
35. Korenberg J, Kawashima H, Pulst S, Et Al. Molecular Definition Of A Region Of Chromosome 21 That Causes Features Of The Down Syndrome Phenotype. J Hum Genet 1990; 47: 236-246.
36. Korenberg Jr, Chen Xn, Schipper R, Et Al. Down Syndrome Phenotypes: The Consequences Of Chromosomal Imbalance. Proc Natl Acad Sci Usa 1994; 91: 4997-5001.
37. Korenberg Jr. Down Syndrome Phenotypic Mapping. En: Epstein Cj, Ed. Morphogenesis And Down Syndrome. New York: Wiley-Liss 1991.
38. Lai F, Williams Rs. A Prospective Study Of Alzheimer Disease In Down Syndrome. Arch Neurol 1989; 46: 849-853.
39. Lejeune J, Gautier M, Turpin R. Etudes Des Chromosomes Somatiques De Neuf Enfants Mongoliens. Comptes Rendues Hebdomadaires Des Seances De Lacademie Des Sciences. Paris. 1959; 248: 602-603.
40. Lemere Ca, Blusztajn Jk, Yamaguchi H, Visniewski T, Saido Tc, Selkow Dj. Sequence Of Deposition Of Heterogeneous Amyloid Beta-Peptides And Apo E In Down Syndrome: Implications For Initial Events In Amyloid Plaque Formation. Neurobiol Dis 1996; 3: 16-32.
41. Mann Dma. Alzheimers Disease And Downs Syndrome. Histopathology 1988; 13: 125-137.
42. Martín Carrasco Manvel. La enfermedad de Alzheimer un trastorno neuropsiquiátrico. Ars Médica 2004 p. 161-184.
43. Mazur-Kolecka B, Kowal D, Sukontasup T, Dickson D, Frackowiak 3. The Effect Of Oxidative Stress On Accumulation Of Apolipoprotein E3 And E4 In A Cell Culture Model Of 8-Amyloid Angiopathy (Caa). Brain Res 2003; 983: 48-57.
44. Mcguire De, Chicoine Ba. Trastornos Depresivos En Los Adultos Con Síndrome De Down. Rev Síndrome Down 1997; 14: 11-16.
45. Mcnamara Mj, Gómez-Isla T, Hyman Bt. Apolipoprotein E Genotype And Deposits Of Abeta40 And Abeta42 In Alzheimer Disease. Arch Neurol 1998; 55: 1001-1004.
46. Patterson, D. The Integrated Map Of Human Chromosome 21. Prog Clin Biol Res 1995.
47. Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, Et Al. Apolipoprotein E, Dementia And Cortical Deposition Of Beta-Amyloid Protein. N Eng J Med 1995; 333: 1242-1247.
48. Prasher Vp, Chowdhury Ta, Rowe Br, Bain Sc. Apoe Genotype And Alzheimers Disease In Adults With Down Syndrome: Meta-Analysis. Am J Ment Retard 1997; 102: 103-110.
49. Prasher Vp, Chung Mc, Haque Ms. Longitudinal Changes In Adaptive Behavior In Adults With Down Syndrome: Interim Findings From A Longitudinal Study. Am J Ment Retard 1998; 103: 40-46.
50. Prasher Vp, Chung Mc. Causes Of Age-Related Decline In Adaptive Behavior Of Adults With Down Syndrome: Differential Diagnoses Of Dementia. Am J Merit Retard 1996; 101: 175-183.
51. Prasher Vp, Farrer Mj, Kessling Am, Et Al. Molecular Mapping Of Alzheimer-Type Dementia In Downs Syndrome. Ann Neurol 1998; 43: 380-383.
52. Prasher Vp, Filer A. Behavioural Disturbance In People With Downs Syndrome And Dementia. J Intellect Disabil Res 1995; 39: 432-436.
53. Prasher Vp. Dementia Questionnaire For Persons With Mental Retardation (Dmr): Modified Criteria For Adults With Down Syndrome. Jappl Res Intellect Disabil 1997; 10: 54-60.
54. Prasher Vp. End-Stage Dementia In Adults With Down Syndrome. Int J Geriat Psychiat 1995; 10:1067-1069.
55. Prasher, V.P. Et Al (1993) Age Of Onset And Duration Of Dementia In People With Down Syndromerintegration Of 98 Reported Cases In The Literature. Internationaljournal Of Geriatric Psychiatry, Vol 8: 915-22
56. Ribes R. El Procés Denvelliment En La Persona Amb La Síndrome De Down. Indicadors Del Procés De Deteriorament Cognitiu I Funcional Relacionats Amb La Demència Alzheimer. Tesis Doctoral, Universidad De Lérida 1999.
57. Schupf N, Kapell D, Lee Jh, Zigman W, Canto B, Tycko B, Mayeux R. Onset Of Dementia Is Associated With Apolipoprotefna E Epsilon 4 In Downs Syndrome. Ann Neurol 1996; 40: 799-801.
58. Schupf N, Patel B, Silverman W, Zigman Wb, Zhong N, Tycko B, Mehta Pd, Mayeux R. Elevated Plasma Amyloid B-Peptide 1-42 And Onset Of Dementia On Adults With Down Syndrome. Neurosci Let 2001; 301: 199-203.
59. Strauss D, Zigman Wb. Behavioral Capabilities And Mortality Risk In Adults With And Without Down Syndrome. Am J Ment Retard 1996; 101: 269-281
60. Teller Jk, Russo C, Debusk Lm, Angelini G, Zaccheo D, Dagna-Bricarelli F, Scartezzini P, Bertolini S, Mann Dm, Yabaton M, Gambetti P. Presence Of Soluble Amyloid Beta Peptide Precedes Amyloid Plaque Formation In Downs Syndrome. Nat Med 1996; 2: 93-95.
61. Van Dyke Dc, Harper Dc, Dyken E. Alzheimers Disease And Down Syndrome. Down Syndrome Quart 1998; 3 (No. 3): 1-11.
62. Vicari S, Carlesimo A, Caltagirone C. Short Term Memory In Persons With Intellectual Disabilities And Down Syndrome. J Intellect Disabil Res 199.5; 39: 532-537.
63. Vicari S, Caselli Mc, Tonucci F. Asynchrony Of Lexical And Morphosyntactic Development In Children With Down Syndrome. Neuropsychol 2000; 38: 634-644.
64. Visser Fe, Aldenkamp Ap, Van Huffelen Ac, Kuilman M, Overweg J, Van Wijk J. Early Signs Of Dementia Checklist. Am J Ment Retard 1997; 101: 400-412.
65. Visser Fe, Kuilman M, Oosting J, Overweg J, Van Wijk J, Van Huffelen Ac. Use Of Electroencephalography To Detect Alzheimers Disease In Downs Syndrome. Acta Neurol Scand 1996; 94: 97-103.
66. Williams Ca, Doms Rw, Lee Vm. Intracellular App Processing And A Beta Production In Alzheimer Disease. Jneuropathol Exp Neurol 1999; 58: 787-794.
67. Wilson Ca, Doms Rw, Lee Vm. Intracellular App Processing And Abeta Production In Alzheimer Disease. Jneuropathol Exp Neurol 1999; 58: 787-794.
68. Zigman Wb, Schupf N, Sersen E, Et Al. Prevalence Of Dementia In Adults With And Without Down Syndrome. Am Jment Retard 1996; 100: 403-412.
69. Zigman Wb, Schupf N, Sersen E, Silverman W. Prevalence Of Dementia In Adults With And Without Down Syndrome. Am J Ment Retard 1995; 100: 403-412.
70. Zigman Wb, Schupf N, Zigman A, Silverman W. Aging And Alzheimer Disease In People With Mental Retardation. En: Bray Nw (Ed), International Review Of Research In Mental Retardation (Vol. 19). New York, Academic Press 1993; P. 41-70.
71. Zigman, W Et Al (1987). Premature Regression Of Adults With Downs Syndrome American Journal Of Mental Deficiency.V.92 N2 161-68.
72. Zitmanova I, Koritar P. Markers of oxidative stress in children with Down syndrome. Clin Chem Lab Med, 2006; 44 (3): 306-10.
* Resumen del trabajo presentado para la obtención del título de Master en Psicogeriaría de la Universiad Autónoma de Barcelona, 7.a edición (2004-206).