Informaciones Psiquiátricas - Segundo trimestre 2008. Número 192
Estado actual del diagnóstico y asesoramiento genético en demencias
Raquel Sánchez-Valle Díaz
Unidad de
Alzheimer y otros trastornos cognitivos.
Servicio de Neurología-Institut Clínic de Neurociències. Hospital Clínic -
Barcelona.
Recepción: 25-02-08 / Aceptación: 26-03-08
RESUMEN
Entre un 1 y un 15% de los casos de demencias neurodegenerativas están determinados genéticamente por la presencia de una mutación en un gen implicado en la patogenia de la enfermedad. En el resto de los casos, los factores genéticos no son determinantes, aunque pueden modificar tanto la susceptibilidad como el fenotipo clínico. En la enfermedad de Alzheimer se han identificado tres genes causales: el gen de la proteína precursora de amiloide (APP) y los genes de las presenilinas 1 (PSEN1) y 2 (PSEN2). La degeneración lobar frontotemporal es, desde el punto de vista genético, heterogénea, pero la mayoría de los casos genéticos son causados por la presencia de mutaciones en el gen de la proteína tau (MAPT) o en el recientemente descrito, gen de progranulina (PGRN). Las enfermedades por priones de origen genético se originan por la presencia de mutaciones en el gen de la proteína priónica (PRNP). La herencia es en todos estos casos del tipo autosómico dominante, pero según el gen implicado la edad de presentación es variable y algunos casos se pueden presentar sin historia familiar. Los programas de asesoramiento genético multidisciplinares permiten diagnosticar, informar y efectuar un seguimiento adecuado al caso concreto en familias afectas de demencias determinadas genéticamente. La identificación de mutaciones causales permite además, efectuar un diagnóstico presintomático en los familiares sanos, pertenecientes a dichas familias, que así lo desean. En este contexto, el asesoramiento genético en familias con enfermedades neurodegenerativas que cursan con demencia es seguro y potencialmente beneficioso.
INTRODUCCIÓN
La demencia es un problema de salud frecuente en la población de países desarrollados, cuya prevalencia global aumenta de forma asociada al envejecimiento de la población. La mayor parte de los casos de demencia son de inicio senil, presentación esporádica y no determinadas genéticamente, aunque el elemento genético (conocido o no) puede tener un papel predisponente o modificador de la enfermedad. Sin embargo, un porcentaje que oscila entre un 1 y un 15% según el tipo de demencia son de origen genético, producidas por una alteración en un gen que interviene en la patogenia de la enfermedad y que se trasmite, habitualmente, con un patón autosómico dominante. Que estos casos genéticamente determinados se presenten con un patrón familiar o no, o a una edad más o menos avanzada va a depender en la mayoría de los casos del gen concreto implicado (y por tanto el perfil clínico con el que éste se presente), hecho que ha de ser tenido en cuenta a la hora de realizar un correcto diagnóstico del caso.
Los avances producidos en los últimos años en la genética molecular de las enfermedades neurodegenerativas ha permitido no sólo proporcionar un diagnóstico genético a los pacientes afectos, sino poder ofrecer además la posibilidad de realizar un estudio predictivo a individuos en riesgo. La realización de este tipo de estudios, en enfermedades para las que no existe un tratamiento preventivo o curativo, no está exenta de riesgos y complicaciones. El diagnóstico de una enfermedad genética no sólo afecta al sujeto que padece el proceso, sino que implica al resto de la familia, y conlleva consecuencias emocionales, éticas, legales, laborales y sociales que han de ser tenidas en cuenta en el proceso diagnóstico. Ante la ausencia de mecanismos legislativos reguladores, los profesionales nos guiamos por recomendaciones elaboradas por comités de expertos. En este sentido, el proceso de asesoramiento y diagnóstico genético en la mayoría de enfermedades neurodegenerativas graves de herencia autosómica dominante se basa en las recomendaciones realizadas para la enfermedad de Huntington, enfermedad neurodegenerativa de origen genético en la que se dispone de una mayor experiencia1.
ASESORAMIENTO GENÉTICO EN DISTINTAS DEMENCIAS DEGENERATIVAS
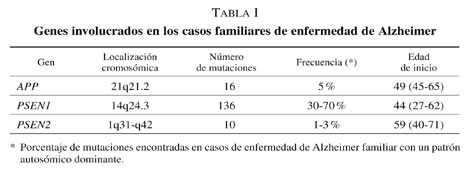
La enfermedad de Alzheimer (EA) afecta a más del 3 % de los sujetos a los 65 años y a 20-30 % de los mayores de 80 años. Las formas de inicio precoz (establecido en un inicio menor a 60 años según algunos autores, menor a 65 años según otros) representan el 1-6 % de los casos y pueden ser de causa genética, habiéndose descrito un patrón de herencia autosómica dominante en un 13 % de ellos2. En los casos genéticos es posible identificar mutaciones patogénicas en tres genes diferentes que intervienen en el procesamiento de la proteína amiloide: la proteína precursora del amiloide (APP)3-4, la presenilina 1 (PSEN1)5-6 y la presenilina 2 (PSEN2)7-8. En los tres casos la penetrancia de la enfermedad en portadores de mutaciones es prácticamente completa (se han descrito algunas excepciones con la PSEN2). Las mutaciones más frecuentes son las asociadas a presenilina 19. En el caso de la APP se han descrito casos de EA asociados, no sólo a mutaciones puntuales, sino a duplicaciones en el gen. Si bien no existen normas establecidas para la realización del estudio genético en la EA, la indicación más clara sería la de un paciente con EA de inicio presenil con historia familiar con patrón autosómico dominante. Para aquellos pacientes de inicio en edad senil podría estar indicado realizar un estudio del gen PSEN2 cuando existan antecedentes familiares con patrón autosómico dominante de enfermedad de inicio presenil en alguno de sus miembros. Para pacientes con EA sin historia familiar conocida (muerte precoz de alguno de sus progenitores o desconocimiento de progenitores biológicos) podría estar indicado realizar un estudio genético cuando la enfermedad hubiese debutado por debajo de los 58 años9. El estudio genético no estaría indicado en casos de inicio senil, aún cuando hubiese historia familiar de la enfermedad, si todos los casos fueran de inicio mayor a 65 años. En la EA de inicio senil, la presencia del alelo «4 en el gen de la apolipoproteína E es el factor de riesgo genético de mayor potencia conocido10. Sin embargo, según las recomendaciones internacionales su estudio no estaría justificado con fines de consejo genético (tabla I).
La degeneración lobar frontotemporal (DLFT) constituye la tercera causa de demencia neurodegenerativa en nuestra población, después de la EA y la demencia por cuerpos de Lewy. La enfermedad se manifiesta inicialmente con trastornos conductuales (demencia frontotemporal), con alteraciones del lenguaje (afasia progresiva no fluente o demencia semántica) o bien con ambas de forma más o menos simultánea o sucesiva, apareciendo posteriormente un deterioro cognitivo más global. En el curso de la enfermedad algunos pacientes desarrollan además parkinsonismo o enfermedad de motoneurona. Entre un 30 y un 40% de los pacientes con DLFT presentan antecedentes familiares de demencia, y un 20% un patrón de herencia autosómica dominante. La DLFT es una entidad clínica, anatomopatológica y genéticamente heterogénea. La mayoría de los casos de origen genético son debidos a la presencia de mutaciones en los genes de proteína asociada a microtúbulos tau (MAPT)11 y de progranulina (PRGN)12-14. Los casos de DLFT asociada a mutaciones en el gen MAPT suponen, según la serie consultada, de un 3 a un 10 % de los casos. Estos casos se presentan habitualmente con la forma conductual de la enfermedad (demencia frontotemporal) y depósito de proteína tau en el estudio anatomopatológico.
Es excepcional su aparición en casos esporádicos y su penetrancia es prácticamente completa a la edad de 65 años15, por lo que la indicación de estudio genético del gen MAPT se suele limitar a los casos de DFT de presentación presenil y familiar. Los casos asociados a mutaciones en el gen PGRN suponen un 5% del total de casos, porcentaje que asciende a un 13% si se consideran sólo los casos familiares16. La presentación clínica es más heterogénea que los casos asociados a mutaciones en el gen MAPT, al igual que la edad de presentación (48 a 83 años), lo que condiciona una penetrancia incompleta en algunas familias y que se hayan descrito casos sin historia familiar (en series recientemente publicadas, hasta un 3% de los casos esporádicos presentan una mutación en PGRN). En el estudio anatomopatológico estos pacientes presentan unas inclusiones intranucleares inmunorreactivas frente a ubiquitina típicas. Las indicaciones de estudio genético de PGRN no están bien establecidas (su relación causal con la DLFT fue descrita por vez primera en julio del 2006), pero posiblemente estaría indicada su realización tanto en casos familiares como en casos esporádicos cuando no se disponga de estudio anatomopatológico.
Las enfermedades por priones son enfermedades neurodegenerativas poco frecuentes, siendo su incidencia aproximada de 2 casos por millón de habitantes y año. La forma más frecuente de enfermedad por priones es la enfermedad de Creutzfeldt-Jakob (ECJ) esporádica, no genéticamente determinada. Estos pacientes presentan un deterioro cognitivo rápidamente progresivo asociado a mioclonias, ataxia y/o parkinsonismo. Las enfermedades por priones o encefalopatías espongiformes transmisibles son enfermedades de declaración obligatoria en España y de su seguimiento se encargan los Servicios de Vigilancia Epidemiológica de cada comunidad.
En Cataluña, un 10% de los casos de enfermedades por priones son de origen genético17, habiéndose descrito casos asociados a las mutaciones puntuales E200K y D178N (ambas en similar frecuencia) y a inserciones en el gen de la proteína priónica (PRNP). Los casos asociados a la mutación D178N, se presentan habitualmente de forma familiar, con una clínica similar a un caso de ECJ o bien con un fenotipo de mayor duración en el que destaca una alteración precoz de los ritmos de sueño, al que se añaden posteriormente deterioro cognitivo, parkinsonismo, ataxia y/o disautonomía y que se conoce con el nombre de Insomnio familiar letal. Los casos asociados a la mutación E200K y alguno de los casos asociados a inserciones en el gen de PRNP presentan un fenotipo y una edad de presentación indistinguible de la forma esporádica. Este hecho, unido a que hasta un 60 % de los casos genéticos asociados a estas mutaciones, se presenten sin historia familiar de enfermedad similar, hace que desde los grupos de estudio europeos se recomiende la realización de consejo genético y estudio genético a todo caso de enfermedad por priones, independientemente de la historia familiar o edad de presentación18.
ASESORAMIENTO GENÉTICO EN DEMENCIAS: PRINCIPOS RECTORES Y NIVELES DE ACTUACIÓN
Los principios éticos de autonomía, beneficencia/no maleficencia, justicia y confidencialidad rigen como a todo acto médico, el proceso de asesoramiento genético en demencias. En este sentido, el principio de autonomía se ha de entender como el derecho del sujeto a decidir, con el conocimiento necesario, si quiere o no quiere proceder a la realización de un estudio genético (derecho a saber, pero también derecho a no saber). Para ello el médico debe informar al individuo de todas las posibles consecuencias derivadas del estudio para que éste pueda tomar libremente una decisión, y resulta conveniente que todo ello quede reflejado en un consentimiento informado. El principio de beneficencia/no maleficencia se ha de respetar poniendo en una balanza en cada paciente concreto los posibles beneficios y perjuicios que se pueden derivar de un estudio genético. El hecho de confirmar el diagnóstico en vida es el principal beneficio de un diagnóstico genético en un individuo enfermo. Aspectos como aliviar la angustia de la incerteza, tomar decisiones sobre planificación familiar, acceder a un tratamiento precoz en un futuro son las principales razones esgrimidas por los sujetos para realizar un estudio genético y avalan su posible efecto beneficioso. Posibles efectos negativas a nivel familiar, social, laboral, pero muy especialmente psicológicas son las principales repercusiones negativas que pueden producirse y que han de procurar ser prevenidas y correctamente tratadas en caso de producirse para garantizar la seguridad del proceso.
Por tanto, el asesoramiento genético es un proceso complejo que incluye, no sólo el análisis genético sino un asesoramiento previo adecuado al paciente y su familia y un seguimiento posterior.
El asesoramiento genético actúa, además, a dos niveles. El primero de ellos sería el de diagnóstico genético de un paciente con demencia. En primer lugar, se habrá de evaluar si un paciente concreto puede o no presentar una demencia determinada genéticamente, considerando el fenotipo clínico, la edad de presentación y los antecedentes familiares de enfermedad similar. Las demencias genéticamente determinadas son habitualmente familiares, pero no siempre, debido a un amplio rango de edad de presentación, ausencia de datos familiares fiables, falsa paternidad o bien por la aparición de mutaciones «de novo». Los siguientes pasos consistirían en valorar si según los conocimientos disponibles podemos identificar el gen/mutación responsable de la enfermedad, plantear qué tipo de estudios genéticos están indicados en el caso y en el caso de llegarse a realizar un análisis genético, interpretar los resultados de las pruebas genéticas, pues la presencia de un cambio genético no siempre se ha de considerar causal, y la ausencia de hallazgos en el estudio genético no siempre puede descartar un origen genético.
El segundo nivel de actuación se encuentra a nivel del sujeto sano, familiar de un paciente con demencia. Cuando el sujeto es familiar de un paciente con demencia de bajo riesgo genético o de alto riesgo pero que no se ha podido realizar un diagnóstico genético que demostrase la presencia de una mutación causal, el asesoramiento genético se limita a realizar una explicación sobre la enfermedad y los riesgos teóricos según la literatura y el caso en concreto. Cuando el sujeto que solicita asesoramiento es familiar de un paciente con demencia genética, con mutación conocida y se encuentra por tanto, a riesgo de ser portador de una mutación causante de demencia degenerativa, el proceso es más complejo. En ese caso, los grupos de expertos recomiendan que el proceso se desarrolle a través de un estricto protocolo multidisciplinar. Para ello, el Hospital Clínic de Barcelona ha creado el programa de información y consejo genético (PICOGEN), que incluye a neurólogos, psiquiatra, psicólogo y genetistas. El programa se inicia con dos visitas de información diferentes con el neurólogo y/o genetista. Si tras estas visitas el sujeto manifiesta su voluntad de realizar el estudio presintomático, que determinará si es portador de la mutación patogénica conocida, éste pasa a ser valorado y asesorado por un psicólogo y un psiquiatra.
Posteriormente, los profesionales implicados en el programa realizan una discusión conjunta del riesgo/beneficio de la realización de la prueba en el individuo concreto. Si especialmente, el psiquiatra o psicólogo consideran que el sujeto no está en condiciones de afrontar un mal resultado, se recomienda el aplazamiento de la prueba y reevaluación pasado un período. Si la evaluación es positiva, se realiza el análisis tras la firma del consentimiento informado.
Todo este procedimiento dura como mínimo tres meses, lo que permite al sujeto realizar las consultas oportunas y darle la oportunidad de cambiar de opinión. Los resultados se dan siempre directamente al sujeto afecto, aconsejándose la presencia de un acompañante o familiar cercano. El programa incluye el seguimiento post-test realizado por un neurólogo, un psicólogo y un psiquiatra, e incluye visitas periódicas, psicoterapia y tratamiento farmacológico si se precisa. El diagnóstico presintomático en estas premisas ha demostrado ser seguro y potencialmente beneficioso19, 20.
BIBLIOGRAFÍA
1. International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Huntingtons Chorea. Guidelines for the molecular genetics predictive test in Huntingtons disease. Neurology. 1994; 44(8): 1533-6.
2. Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999; 65: 664-70.
3. Glenner GG, Wong CW. Alzheimers disease and Downs syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 1984; 122: 1131-5.
4. Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidabi L et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimers disease. Nature 1991; 349: 704-706.
5. Schellenberg GD, Bird TD, Wijsman EM, Orr HT, Anderson L, Nemens E, et al. Genetic linkage evidence for a familial Alzheimers disease locus on chromosome 14. Science. 1992; 258: 668-71.
6. Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimers disease. Nature. 1995; 375: 754-60.
7. Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, et al. Familial Alzheimers disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimers disease type 3 gene. Nature. 1995; 376: 775-8.
8. Bird TD, Levy-Lahad E, Poorkaj P, Sharma V, Nemens E, Lahad A et al. Wide range in age of onset for chromosome 1-related familial Alzheimers disease. Ann Neurol 1996; 4: 932-6.
9. Lleó A, Blesa R, Queralt R, Ezquerra M, Molinuelo JL, Peña-Casanova J et al. Frequency of mutacions in the presenilin and amyloid precursor protein genes in early-onset Alzheimer disease in Spain. Arch Neurol 2002; 59: 1759-1763.
10. Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimers disease. Lancet. 1993 Sep 18; 342(8873): 697-9.
11. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998; 393: 702-705.
12. Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006; 442: 916-9.
13. Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006; 442: 920-4.
14. Lleó A, Sánchez-Valle R, Reñé R, Ezquerra M, Rey MJ, Tolosa E, Ferrer I, Molinuelo JL Late-onset frontotemporal dementia associated with a no-vel PGRN mutation. J Neural Transm 2007 Apr 10; [Epub ahead of print].
15. Stanford PM, Brooks WS, Teber ET, Hallupp M, McLean C, Halliday GM, et al. Frequency of tau mutations in familial and sporadic frontotemporal dementia and other taupathies. J Neurol 2004: 251; 1098-104.
16. Le Ber I, van der Zee J, Hannequin D, Gijselinck I, Campion D, Puel M, Laquerriere A, De Pooter T, Camuzat A, Van den Broeck M, Dubois B, Sellal F, Lacomblez L, Vercelletto M, Thomas-Anterion C, Michel BF, Golfier V, Didic M, Salachas F, Duyckaerts C, Cruts M, Verpillat P, Van Broeckhoven C, Brice A. Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat. 2007 Apr 13; [Epub ahead of print].
17. Sánchez-Valle R, Nos C, Yagüe J, Graus F, Domínguez A, Saiz A, for the Catalan Collaborative Study Group for CJD. Clinical and genetic features of human prion diseases in Catalonia. Eur J Neurol. 2004 Oct; 11(10): 649-55.
18. Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, Collins SJ, Boyd A, Giulivi A, Coulthart M, Delasnerie-Laupretre N, Brandel JP, Zerr I, Kretzschmar HA, de Pedro-Cuesta J, Calero-Lara M, Glatzel M, Aguzzi A, Bishop M, Knight R, Belay G, Will R, Mitrova E; EUROCJD. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005 Nov; 118(2): 166-74.
19. Molinuello JL, Pintor L, Peri JM, Lleó A, Oliva R, Marcos T, et al. Emotional reactions to predictive testing in Alzheimers disease and other inherited dementias. Am J Alz Dis 2005; 20: 233-8.
20. Steinbart EJ, Smith CO, Poorkaj P, Bird TD. Impact of DNA testing for early-onset familial Alzheimer disease and frontotemporal dementia. Arch Neurol. 2001; 58: 1828-31.